Mechanism explainer
Adenosine and Sleep Pressure: The Molecular Science of Why You Get Tired
Explains how adenosine — a metabolic byproduct of neuronal energy use — accumulates during wakefulness, binds to distinct receptor subtypes to suppress arousal and gate sleep, and why only slow-wave sleep fully resolves this pressure; covers the dissociable roles of A1 and A2A receptors, the mechanics of the caffeine mask, and what chronic caffeine use does to the adenosine system.
The Molecular Event Behind That Afternoon Heaviness
Around mid-afternoon, something shifts. Your eyes lose focus. Your thinking slows. You reach for coffee. That experience has a precise molecular explanation — not a vague sense of fatigue, but a measurable chemical event happening in specific regions of your brain.
Every time a neuron fires, it consumes adenosine triphosphate (ATP), the cell's primary energy currency. As ATP is broken down to power that activity, it sheds phosphate groups — becoming ADP, then AMP, and finally adenosine. That adenosine is released into the space between cells, where it begins to accumulate. The longer your neurons have been active, the more adenosine has built up around them.
Adenosine is not a sleep hormone your brain manufactures on a schedule. It is the spent-energy marker of neural work — an inevitable byproduct of staying awake and thinking. Understanding this distinction changes how you interpret tiredness entirely.
What Adenosine Is: The Brain's Spent-Energy Marker
The brain is the most metabolically demanding organ in the body, consuming roughly 20% of the body's total energy despite representing only about 2% of its mass. Neurons require continuous ATP to maintain ion gradients, transmit signals, and support the biochemical machinery of cognition. This energy demand never fully stops during wakefulness.
The pathway from energy use to sleep pressure runs through a short enzymatic sequence. ATP breaks down to ADP during energy transfer, then to AMP as more phosphate groups are shed. AMP is then converted to adenosine by a class of enzymes called 5'-ecto-nucleotidases, which are present on the outer surface of neurons and glial cells. The adenosine that results is released into the extracellular space, where it can bind to receptors on nearby cells.
- ATP (adenosine triphosphate) — the cell's energy currency, used to power neural firing
- ADP (adenosine diphosphate) — formed as ATP releases one phosphate group during energy transfer
- AMP (adenosine monophosphate) — formed as ADP releases another phosphate group
- Adenosine — the final product, released extracellularly via 5'-ecto-nucleotidase activity
This means that every cognitive task, every sensory experience, every moment of sustained attention generates a small increment of adenosine. Sleep pressure is not imposed from outside — it is produced by the brain's own activity. The more demanding your waking hours, the more adenosine accumulates.
Where Adenosine Accumulates — and How
Adenosine does not rise uniformly across the entire brain. The two regions with the strongest documented accumulation during wakefulness are the basal forebrain and the cortex — both areas densely involved in arousal, attention, and cognitive processing. The basal forebrain is particularly important because it contains the cholinergic neurons that actively promote wakefulness; adenosine's effect on these cells is a direct suppression of arousal.
A key mechanism behind this accumulation involves astrocytes — the star-shaped glial cells that outnumber neurons and play a supporting metabolic role throughout the brain. Astrocytes release ATP through a process called gliotransmission, triggered by calcium signals that rise in proportion to surrounding neural activity. Extracellular enzymes then degrade that released ATP into adenosine, adding to the pool of sleep-promoting signal in the local environment.
A subtler point concerns the kinetics of adenosine accumulation. The intuitive model — adenosine rising slowly and steadily across the entire waking day like sand filling an hourglass — is an oversimplification. Research reviewed by Reichert, Deboer, and Landolt (2022) suggests that under normal undisturbed sleep-wake conditions, adenosine in the brain follows more of a square-wave pattern: an abrupt rise at the transition from rest to the active phase, followed by relative stability during consolidated wakefulness. The dramatic linear buildup model applies most clearly when wakefulness is extended well beyond its normal daily duration — during sleep deprivation, for instance. This nuance matters for interpreting why moderate sleep restriction compounds over days rather than producing a single catastrophic crash.
How Adenosine Creates Sleep Pressure: A1 and A2A Receptors Do Different Jobs
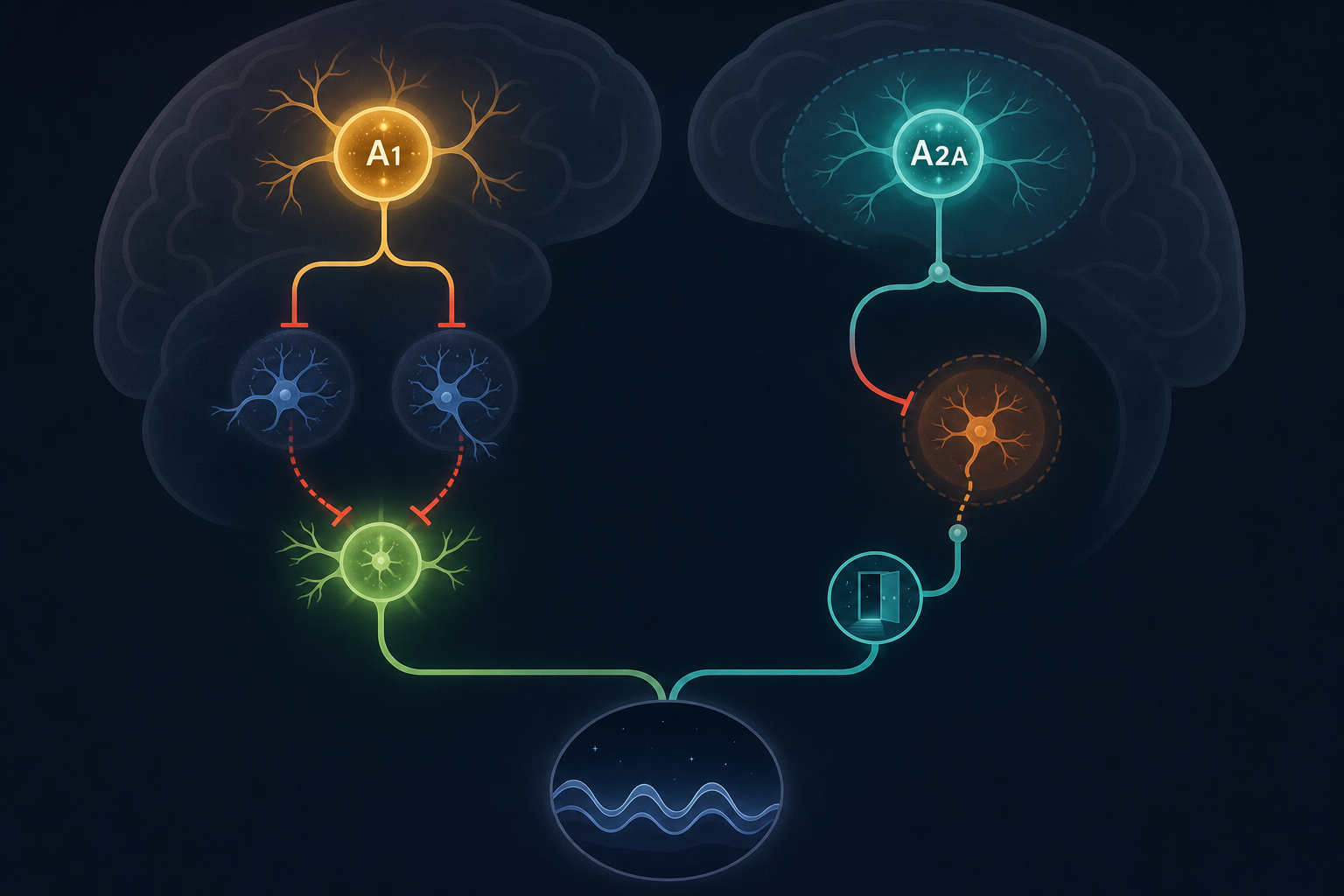
Adenosine does not create sleep pressure through a single pathway. It acts on at least two distinct receptor subtypes — A1 and A2A — that operate in different brain regions and perform dissociable functions. Understanding this distinction is essential to understanding why caffeine works the way it does, and why sleep need and sleep gating are not the same thing.
A1 Receptors: Modulating the Depth and Resolution of Sleep Need
A1 receptors are distributed broadly across the brain, with high concentrations in the basal forebrain, cortex, hippocampus, and brainstem. When adenosine binds to A1 receptors in the basal forebrain, it suppresses the cholinergic neurons that actively promote wakefulness and cortical arousal. It also inhibits histaminergic neurons in the tuberomammillary nucleus (TMN) of the hypothalamus — the same neurons that the antihistamine diphenhydramine targets — and suppresses orexin/hypocretin neurons in the lateral hypothalamus, which are critical for maintaining stable arousal.
The net effect of A1 receptor activation is a broad suppression of arousal-promoting circuits. Simultaneously, A1 receptor activity disinhibits the ventrolateral preoptic area (VLPO), a cluster of neurons in the hypothalamus that actively promote sleep. When arousal circuits are suppressed and the VLPO is released from inhibition, the brain tips toward sleep.
Critically, A1 receptors appear to mediate the homeostatic dimension of sleep need — the accumulation of sleep debt and the intensity of slow-wave activity during recovery. When A1 receptors are conditionally knocked out in forebrain glutamatergic neurons in animal models, the slow-wave activity rebound after sleep deprivation is abolished, even though the animals still fall asleep. This confirms that A1Rs are essential for expressing and resolving sleep need, not just for inducing sleep onset.
A2A Receptors: Gating Sleep Onset via the Nucleus Accumbens
A2A receptors are concentrated in the nucleus accumbens shell — a region more commonly associated with reward processing than sleep. Their role in sleep was clarified through chemogenetic and optogenetic experiments showing that activating A2A receptor-expressing neurons in the nucleus accumbens shell produces robust slow-wave sleep, while inhibiting them blocks sleep induction without affecting the homeostatic sleep rebound.
The mechanism involves the ventral pallidum, an arousal-promoting structure downstream of the nucleus accumbens. When adenosine activates A2A receptors in the NAc shell, it inhibits the ventral pallidum, removing an active brake on sleep-promoting circuits. This is a gating function: A2A receptors allow the brain to enter sleep state, rather than modulating how much sleep pressure has accumulated.
The distinction matters practically. A1 receptors govern how much sleep need has built up and how deeply it resolves during recovery. A2A receptors govern whether the brain can transition into sleep at all. Caffeine's primary wake-promoting effect — as confirmed by studies in A2A receptor knockout mice, which are largely unaffected by caffeine's arousal-promoting properties — operates through A2A receptor blockade in the nucleus accumbens shell.
| Receptor | Primary Location | Function | Effect of Adenosine Binding | Caffeine Relevance |
|---|---|---|---|---|
| A1 | Basal forebrain, cortex, hippocampus, TMN, lateral hypothalamus | Modulates depth and resolution of homeostatic sleep need | Suppresses cholinergic, histaminergic, and orexin arousal neurons; disinhibits VLPO sleep-promoting neurons | Blockade contributes to secondary arousal effects; A1R upregulation drives caffeine tolerance |
| A2A | Nucleus accumbens shell | Gates sleep onset (allows brain to enter sleep state) | Inhibits ventral pallidum, removing brake on sleep-promoting circuits; produces robust slow-wave sleep | Primary target of caffeine's wake-promoting effect; A2A knockout mice are largely insensitive to caffeine's arousal action |
Adenosine as the Substrate of Process S
In Alexander Borbély's influential two-process model of sleep regulation, sleep need is governed by two interacting forces: Process S, a homeostatic accumulator that rises during wakefulness and falls during sleep, and Process C, the circadian pacemaker that sets the timing of sleep and wake windows. Adenosine is the leading neurochemical candidate for the biological substrate underlying Process S.
The measurable readout of Process S in the sleeping brain is slow-wave activity (SWA) — EEG oscillations in the 0.5–4.5 Hz range that dominate NREM stage 3 (N3) sleep. SWA magnitude at the start of a sleep period reflects how much homeostatic debt has accumulated; it declines exponentially across the night as the debt is resolved. After sleep deprivation, SWA rebounds sharply — the brain's way of clearing a larger-than-normal adenosine load.
How Sleep Clears the Debt: Slow-Wave Sleep and Adenosine Kinase
The clearance of adenosine during sleep is not passive diffusion — it is an active enzymatic process. Two enzymes are primarily responsible:
- Adenosine kinase phosphorylates adenosine back to AMP, effectively recycling it into the ATP pathway. This enzyme is expressed predominantly in astrocytes, not neurons — which means glial metabolism drives both the accumulation and the clearance of sleep-regulatory adenosine.
- Adenosine deaminase converts adenosine to inosine, an irreversible degradation product that does not bind adenosine receptors. This pathway provides a secondary clearance route.
Both pathways are most active during slow-wave (N3) sleep. This is why N3 is not simply the deepest stage of sleep — it is the physiologically functional stage where homeostatic debt is resolved. Stages N1 and N2, and REM sleep, do not provide equivalent adenosine clearance. This has a direct implication: any factor that fragments or suppresses N3 sleep leaves adenosine debt partially unresolved, regardless of how many total hours were spent in bed.
The magnitude of SWA in recovery sleep scales proportionally with prior debt. A single night of total sleep deprivation produces a sharp SWA rebound in the first recovery sleep period. Partial sleep restriction over multiple nights produces a more gradual but cumulative SWA elevation during recovery. The brain tracks what it is owed.
Caffeine: A Receptor Blocker, Not an Energy Donor
Caffeine is the world's most widely consumed psychoactive substance, and the mechanism behind its alerting effect is frequently misunderstood. Caffeine does not give you energy. It does not replenish ATP or accelerate neural metabolism. What it does is act as a competitive antagonist at adenosine receptors — it occupies the same binding sites that adenosine would use, without activating them.
Crucially, caffeine's presence does not stop adenosine from being produced. ATP breakdown continues at the same rate. Adenosine continues to accumulate in the extracellular space. What caffeine does is prevent that accumulated adenosine from signaling — the receptors are occupied, the downstream suppression of arousal circuits does not occur, and you remain alert. But the debt is still building in the background.
Caffeine has roughly equal affinity for both A1 and A2A receptors at normal doses. However, its primary wake-promoting effect operates through A2A receptor blockade in the nucleus accumbens shell — the same gating mechanism described above. This was confirmed in A2A receptor knockout mice, which showed substantially reduced sensitivity to caffeine's arousal-promoting effects compared to wild-type animals.
There is also a secondary interaction worth noting. Adenosine modulates how the suprachiasmatic nucleus (SCN) — the brain's circadian pacemaker — responds to light. Research by Jagannath et al. (2021) demonstrated that adenosine encodes sleep history and that this signal influences circadian entrainment: under conditions of elevated adenosinergic tone (such as sleep deprivation), adenosine attenuates light-induced phase shifts of the circadian clock. Caffeine's blockade of adenosine receptors in the SCN therefore adds a circadian dimension to its effects beyond pure sleep pressure masking — a secondary consideration relevant to understanding why late caffeine intake can shift your internal timing, not just delay your sleep onset. For the broader picture of how light and circadian timing interact, see the article on how light affects sleep and circadian rhythm.
Chronic Caffeine Use and Receptor Adaptation
Habitual caffeine consumers often report that their morning coffee no longer makes them feel more alert than they do on days without it — it simply prevents them from feeling worse. This is not a placebo effect. It reflects a genuine biological adaptation in the adenosine system.
Animal studies show that chronic caffeine exposure increases plasma adenosine levels and upregulates A1 receptor density — the brain compensates for persistent receptor blockade by making more receptors. The result is that the baseline adenosinergic tone rises, and the habitual consumer needs caffeine not to exceed their normal alertness level, but simply to return to it.
Recovery sleep after extended wakefulness restores elevated A1 adenosine receptor availability in the human brain. Sleep deprivation resulted in a higher A1AR availability... The increase that was observed after 52 hours of wakefulness was restored to control levels during a 14-hour recovery sleep episode.
This finding from Elmenhorst et al. (2017), using PET imaging with a selective A1 receptor tracer in humans, has two important implications. First, it provides direct human evidence that the adenosine system is dynamically regulated by sleep history — A1 receptor availability rises with sleep deprivation and normalizes with recovery sleep. Second, it revealed individual differences: people who showed larger A1 receptor availability increases after sleep deprivation were more resilient to cognitive impairment from that deprivation, suggesting that the adenosinergic system's plasticity partly explains why some people tolerate sleep loss better than others.
What happens to sleep architecture under chronic daytime caffeine use? A 10-day randomized crossover trial by Weibel et al. (2021) in habitual male consumers found that taking 150 mg of caffeine three times daily — with the last dose more than eight hours before bedtime — did not significantly alter slow-wave activity or slow-wave sleep duration compared to placebo. The homeostatic clearance mechanism appears largely intact under these conditions.
What This Means in Practice
The molecular architecture of adenosine sleep pressure has several implications that are not obvious from the surface-level "adenosine makes you sleepy" framing.
- Cutting sleep short does not eliminate adenosine debt. Waking up after six hours when your body needed eight does not reset the system. The unresolved adenosine remains, the A1 receptor suppression of arousal circuits persists, and the next day begins with a deficit that compounds if the pattern repeats.
- The caffeine crash is a debt-rebound event, not an energy depletion. When caffeine clears its receptor binding sites (its half-life is approximately 5–6 hours in most adults), the adenosine that accumulated during those hours floods previously blocked receptors simultaneously. The crash is proportional to the masked debt.
- Habitual caffeine consumers experience tolerance, not enhanced alertness. Receptor upregulation means the adapted adenosine system requires caffeine as a baseline condition. Alertness relative to a non-adapted baseline is not reliably improved; the caffeine restores what adaptation has shifted downward.
- Only slow-wave sleep fully resolves homeostatic pressure. A nap that stays in N1 and N2 provides some benefit but does not activate adenosine kinase at the same rate as N3. Sleep fragmented by arousal events — from apnea, noise, alcohol-induced suppression, or aging-related changes — leaves the clearance mechanism partially engaged.
- Adenosine cannot be supplemented. Unlike melatonin, adenosine cannot cross the blood-brain barrier in meaningful amounts, and even if it could, its vasodilatory effects would create serious cardiovascular complications. There is no supplement equivalent of the adenosine clearance that slow-wave sleep provides.
One practical comparison worth making: diphenhydramine, the antihistamine found in most OTC sleep aids, promotes sedation by blocking histamine H1 receptors on the same tuberomammillary nucleus neurons that adenosine suppresses via A1 receptors. This means diphenhydramine is acting downstream of adenosine — it mimics part of what accumulated adenosine would do to arousal circuits, but without engaging the homeostatic machinery that makes sleep restorative. For a detailed look at how that mechanism plays out clinically, including tolerance, next-day grogginess risk, and population safety considerations, see the article on diphenhydramine as a sleep aid.
Supports these guides
Spot an error or have clinical feedback?
Because this article covers clinical, medication, or safety information, we use a moderated correction channel instead of open public comments. Let us know if something about “Adenosine and Sleep Pressure: The Molecular Science of Why You Get Tired” needs a closer look.
Send feedback on this article